 By Laura Fonda Hochnadel
By Laura Fonda Hochnadel
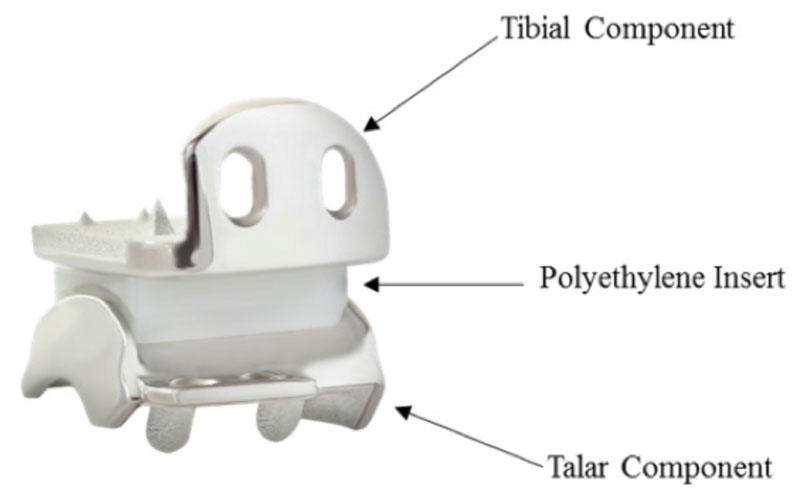
The Hintermann Series H3 Total Ankle Replacement System (DT MedTech, Towson, MD), received premarket approval from the US Food & Drug Administration (FDA). This is a non-cemented, implanted medical device intended to replace a painful arthritic ankle joint and improve ankle motion and is indicated for use in the first surgical treatment for the joint. It is comprised of 3 components: the tibial component, which covers the very bottom of the tibia/upper portion of the ankle joint; the talar component, which covers the lower bone of the ankle joint, the talus; and the polyethylene sliding insert that is placed between the two. It offers 4 degrees of freedom, providing an unconstrained tibial articulating surface and a semi-constrained talar articulating surface.
The H3 Tibial and Talar components are available in size 1 to 6, in right and left versions. The inlay is available in size 1 to 6, in 4 thicknesses (5, 6, 7, and 9 mm). The Flat Cut Talar Component is available in size 1 to 5, in right and left versions. It has the same design features of the Talar Component; however, the thickness has been increased to compensate for the additional bone resection, if needed based on patient anatomy.
In the clinical data provided to the FDA, patients who received the Hintermann Series H3 Total Ankle Replacement System (H3) had fewer serious device-related adverse events (other than a revision or removal) 2 years after surgery compared with another similar total ankle replacement system. Additionally, the clinical data used to support the premarket approval shows 95.9% of patients who received the implant were moderately to very satisfied with the procedure at 5 years post-implantation, and survivorship of 88% at 7 years. The H3 has been implanted in more than 20,000 patients in markets outside the US since May 2000.
 Contraindications include skeletal immaturity; poor quality bone that keeps the bone from supporting the implant; current or past infection in the ankle joint or adjacent bones; poor ankle or knee alignment, poor support from ankle ligaments, or severe deformity of the ankle or nearby bones and joints, which can keep the foot from being flat to the ground; neuropathic arthropathy; poor muscle function around the ankle; insufficient blood flow to the foot bones; poor skin and soft tissue quality around the surgical area; immunosuppressive therapy; prior ankle fusion or a previous total ankle replacement; high demand sport activities; and suspected or documented metal allergy or intolerance.
Contraindications include skeletal immaturity; poor quality bone that keeps the bone from supporting the implant; current or past infection in the ankle joint or adjacent bones; poor ankle or knee alignment, poor support from ankle ligaments, or severe deformity of the ankle or nearby bones and joints, which can keep the foot from being flat to the ground; neuropathic arthropathy; poor muscle function around the ankle; insufficient blood flow to the foot bones; poor skin and soft tissue quality around the surgical area; immunosuppressive therapy; prior ankle fusion or a previous total ankle replacement; high demand sport activities; and suspected or documented metal allergy or intolerance.
Toe Fixation System Receives 501K Clearance from FDA
The BOSS Toe Fixation System (Arthrosurface, Franklin, MA) has received 510K clearance from the US Food & Drug Administration (FDA). This product is intended for use in the treatment of patients with arthritis of the first metatarsalphalangeal (MTP) joint in the presence of good bone stock along with the following conditions: hallux valgus, hallux rigidus, and an unstable or painful MTP joint. The device is a motion-preserving alternative to toe fusion; it stabilizes the joint while preserving the length and mechanical access of the toe.
The BOSS is a single-use implant that can be used with or without bone cement. Fixation components are being included in the system that contain an additional ring of material intended to improve stabilization in a first metatarsal that presents with a distal bone void.
A 510(K) is a premarket submission made to the FDA that demonstrates the device is at least as safe and effective when compared to a legally marketed device that is not subject to premarket approval. In this case, the BOSS Toe Fixation system is demonstrated to be substantially equivalent to the company’s HemiCAP system.
 Meniscus Implant Receives FDA Breakthrough Device Designation
Meniscus Implant Receives FDA Breakthrough Device Designation
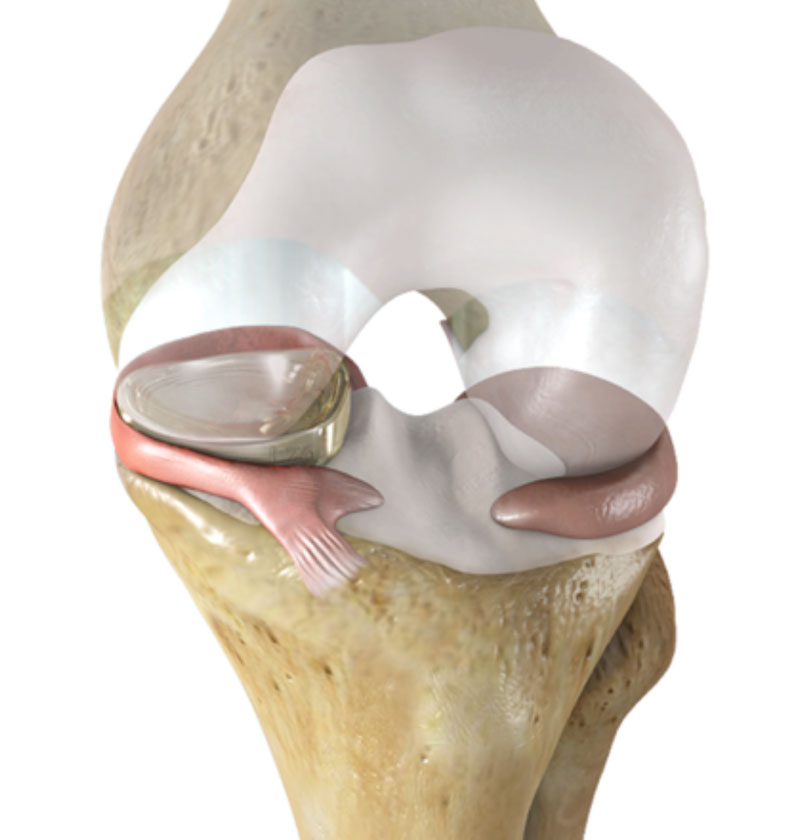
The NUsurface Meniscus Implant (Active Implants, Memphis, TN), has received a breakthrough device designation from the US Food and Drug Administration (FDA). The company said it is the first artificial meniscus to be marketed in Europe and, if cleared by the FDA, would be the first artificial meniscus in the US. The FDA Breakthrough Devices Program was implemented to expedite the development and review process for medical devices that are novel or offer new technology for patients with life-threatening or irreversibly debilitating conditions. This program is designed to ensure patients and healthcare providers have more timely access to vital devices.
The NUsurface Meniscus Implant is an investigational treatment for patients with persistent knee pain following medial meniscus surgery. It is intended to fill the gap in treatment options between minimally invasive meniscus repair and total knee replacement. The artificial meniscus is made from medical-grade polymer and—as a result of its materials, composite structure, and design—does not require fixation to bone or soft tissues. The implant mimics the function of the natural meniscus and redistributes loads transmitted across the knee joint.
Laura Fonda Hochnadel is Associate Editor for Lower Extremity Review.