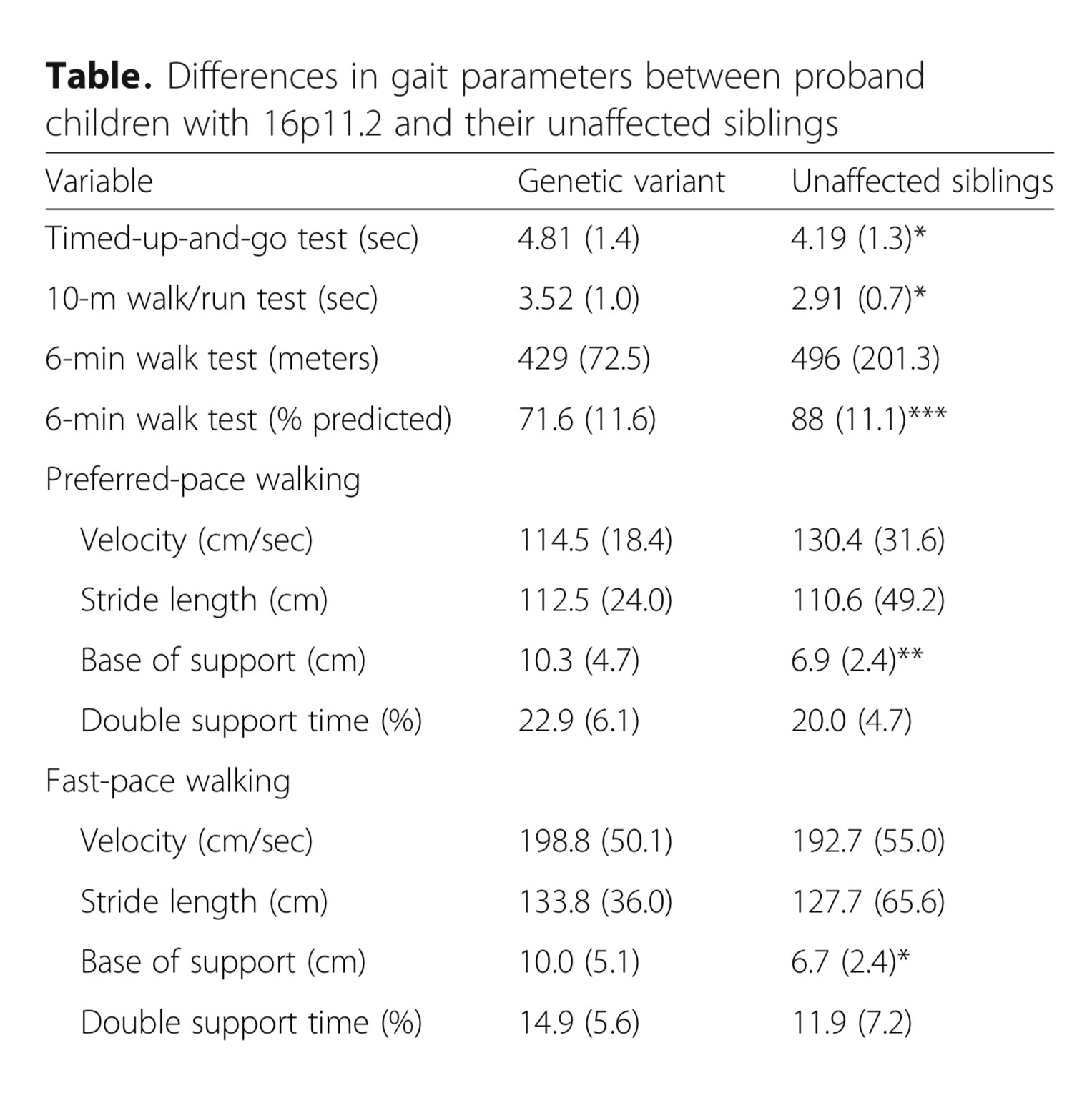
Significant differences in gait parameters between children with and without genetic variant 16p.11.2. Descriptive values are medians (interquartile range) Abbreviations: m meter, min minute, cm centimeters, sec seconds p < 0.05 (*), p < 0.01 (**), p < 0.001(***)
Neurodevelopmental disorders are frequently associated with motor impairments including locomotion. These study findings highlight the importance of using precise measures to differentiate motor dysfunction in neurodevelopmental disorders.
By Sylvie Goldman, Aston K. McCullough, Sally Dunaway Young, Carly Mueller, Adrianna Stahl, Audrey Zoeller, Laurel Daniels Abbruzzese, Ashwini K. Rao, and Jacqueline Montes.
Neurodevelopmental disorders are characterized by a wide range of impairments with broad severity level. Among the affected developmental functions, motor impairments represent the earlier and most visible signs. Yet, the motor phenotypes are often difficult to quantify precisely in children with neurodevelopmental disorders especially when accompanied by high behavioral comorbidities such as those with autism spectrum disorder (ASD) who may display low compliance, distractibility, and poor imitation skills. These behavioral challenges often confound traditional command-based motor assessments. With recent technological advances, many gene-based syndromes have been identified that provide ways to reduce phenotypic heterogeneity.
Focusing on a homogenous cohort of children with selected genetic conditions such as 16p11.2, the most frequent etiologies for ASD have led to fruitful genotype/phenotype findings. Children with 16p11.2 mutation (deletion or duplication) present with a range of neurodevelopmental impairments affecting mostly cognitive (ie, language) and behavioral (attention deficit, autism spectrum disorder) and motor (ie, coordination) functions. The prevalence of comorbid diagnosis of autism spectrum disorder in individuals with 16p11.2 deletion has been estimated between 20 and 33%. Speech articulation, limb and trunk hypotonia, abnormalities of agility, and seizures have been reported in a large cohort of carriers with both 16p11.2 deletion and duplication. In addition to the neurological phenotypes, studies have reported an unexplained high prevalence of obesity, which may be related to level of physical activity. Despite the high prevalence of motor impairment in children with 16p11.2 with and without ASD, the majority of studies so far have relied on parent questionnaires to study the motor profile.
In this descriptive study, we used an instrumented walkway and a battery of standardized quantitative measures of locomotion (gait) and mobility (functionality) to compare 16p11.2 probands with and without a diagnosis of ASD to a group of non-affected siblings.
Methods
Children included in this study were recruited as part of a large research family meeting that provided the genetic (16p11.2 deletion/duplication) and behavioral diagnosis (ASD) based on prior evaluations. Thirty-six children (21 probands with 16q11.2 mutation, deletion (n = 18), or duplication (n = 3) and 15 unaffected siblings), with a mean age of 8.5 years (range 3.2–15.4), 55% male, and 23% incidence of ASD, completed the study. Gait assessments included 6-min walk test (6MWT), 10-m walk/run test (10MWR), timed-up-and-go test (TUG), and spatio-temporal measurements of preferred- and fast-paced gait. Children were required to walk in 10-m bouts across the GAITRite™ instrumented walkway to calculate temporal and spatial gait parameters (velocity and stride length). Measures of balance using percent time in double support (%) and base of support (cm) from the walkway were collected during these assessments. Subsequently, gait data from the 6 children diagnosed with ASD were compared with 6 age-matched non-ASD genetic variant and 6 non-ASD sibling controls.
The Pediatric Evaluation of Disability Inventory-Computer Adaptive Test (PEDI-CAT), a caregiver-questionnaire, was administered as a measure of daily living motor functionality. The PEDI-CAT is comprised of 3 functional domains: Daily Activities (eg, eating, dressing), Mobility (eg, standing, running both assessed in home and school environments), and Social/Cognitive (eg, communication, interaction, problem solving), and an additional Responsibility domain related to the extent to which the child is able to seek assistance as needed and direct others to enable independent living.
Full statistical analyses are detailed in the original text. Preliminary analyses revealed that gait parameters tended to be non-normally distributed in this sample. Resultantly, differences in gait parameters between the 2 groups were tested using a Wilcoxon rank sum test, and the rank sum test statistic (W) was reported. The Wilcoxon rank sum test is appropriate for detecting differences in medians between 2 independent samples, especially when data do not follow a normal distribution.
Results
Proband children had longer performance times on the TUG (W= 408; P = 0.009) and 10MWR (W= 460; P = 0.02) and had lower percent of predicted distances (ie, walked shorter distance) on the 6MWT (W= 230; P < 0.001) than children without the genetic variant. As shown in the Table, probands had a wider base of support in both walking conditions (W= 478; P = 0.004). Probands had significantly lower scores in all domains of the PEDI-CAT, including the Mobility subscale compared to siblings (W= 287; P = 0.001). In proband children, caregiver-reported scores for Daily Activity subscale were positively associated with the 6MWT ( ρ = 0.49, P < 0.05). There was no association in proband children and siblings between caregiver reported scores on PEDI-CAT subscales (Daily Activities, Mobility, Social/Cognitive, and Responsibility) and other quantitative measures of gait (6MWT percent of predicted distance, 10MWR, and base of support) and function (TUG) (P > 0.05).
Discussion
For the first time, this study reports the results of a detailed battery of gait assessments to characterize motor performance in children with 16p11.2 deletion or duplication. We found that probands have reduced performance on functional motor tasks and lower endurance compared to their non-affected siblings. Despite the high prevalence of motor comorbidities in 16p11.2 probands, so far, studies have used parent report or global functional measures to assess motor profiles. However, parental reports do not provide detailed characterization of motor performance and lack sensitivity to changeneeded for outcome studies or clinical trials.
Here, we used a comprehensive protocol to compare gait and balance in probands and their unaffected siblings. We identified differences in markers of balance and standardized functional assessments. Children with 16p11.2 mutation demonstrated impaired balance and slower speed during walking and running tasks. Reduced performance on clinical assessments was also identified in parent reports of lower functional abilities using standardized questionnaires. Furthermore, reduced endurance was associated with parents’ responses about their child’s daily activities performance. This study provides initial support of these assessments for use in longitudinal natural history studies and clinical trials in 16p11.2.
Detailed characterization of gait is necessary to quantify the severity and the trajectory of the motor impairments. More importantly, it sheds light on motor pathways that may be involved in known copy number variant genetic conditions such as 16p11.2, the most frequent genetic etiology of ASD. In view of the significant heterogeneity in the cognitive, behavioral, and motor phenotype of children with neurodevelopmental disorders including ASD, the identification of quantifiable features to differentiate their motor phenotypes is highly valuable. Furthermore, this approach may prove useful in new genotype-phenotype studies using motor performance as a target.
Impaired locomotion and possibly obesity which was commonly reported among the 16p11.2 group relative to their unaffected siblings may influence their engagement in a range of physical activities and in turn affect their social life and wellness. Furthermore, recent studies focusing on interactions between motor functions and social development in neurodevelopmental disorders highlight increased screen time, obesity, and sedentary lifestyles predisposing these children to other behavioral comorbidities. However, our results using the PEDI-CAT, a useful parent report to measure daily activity function including social and cognitive abilities, may not necessarily capture the entire social domain. In order to better understand the influence of motor function on social development in this population, a more detailed and direct assessment of social skills is needed.
Yet, in general, developmental studies point to the benefit of engaging children with motor and cognitive impairments in physical activities in an adaptive way to allow them to thrive socially and physically. Future studies in 16p11.2 and other related disorders will need to be designed to assess more precisely how the motor profile affects the level of physical activity and the risk for these comorbidities.
We recognize the following limitations of this study, including a small convenience sample size of selected families and the use of siblings as control groups instead of unrelated typically developing children. The small subgroup of six probands with a confirmed diagnosis of ASD was not large enough to examine the specific relationship between ASD and motor function; however, it was consistent with the reported 30% prevalence of ASD in 16p11.2.
Conclusions
Our study reports quantitative gait measures together with parents’ measures of functionality and as such provides novel characterization of the motor impairments in children with 16p11.2. These findings demonstrate the applicability of our protocol and support its utility to identify and define motor dysfunction in children with neurodevelopmental disorders despite their behavioral challenges.
This article has been excerpted from “Quantitative gait assessment in children with 16p11.2 syndrome,” by the same authors, which appeared in the Journal of Neurodevelopmental Disorders; 2019;11:26). doi.org/10.1186/s11689-019-9286-9. Minor editing has occurred and references have been removed for brevity. Use is per the Creative Commons Distribution 4.0 International License. To read the full article, go to https://jneurodevdisorders.biomedcentral.com/articles/10.1186/s11689-019-9286-9