![1diabetesFigure_3a[1]](https://lermagazine.com/wp-content/uploads/2013/01/1diabetesFigure_3a1.jpg)
Prevention and treatment of foot ulcers in patients with diabetes requires an understanding of the various factors that contribute to increased risk, including anatomical deformity, poor vascular function, and diminished capacity for healing at a microscopic level.
By Allyson Berglund, DPM, Matthew Juriga, DPM, Aristidis Veves, MD, DSc, and Thanh Dinh, DPM
The worldwide prevalence of diabetes, estimated as 2.8% in 2000, is projected to increase to 4.4% in 2030;1 as many as 15% of the individuals diagnosed with diabetes will develop a foot ulceration during their lifetime.2 Therefore, as the incidence of diabetes continues to rise, foot ulcerations will follow. Foot ulceration is known to prolong hospital stays and also carries with it an increased risk of amputation and mortality.3,4 Understanding the etiology of foot ulceration is essential for effective ulcer evaluation, treatment, and prevention of subsequent complications.
The pathway to foot ulceration
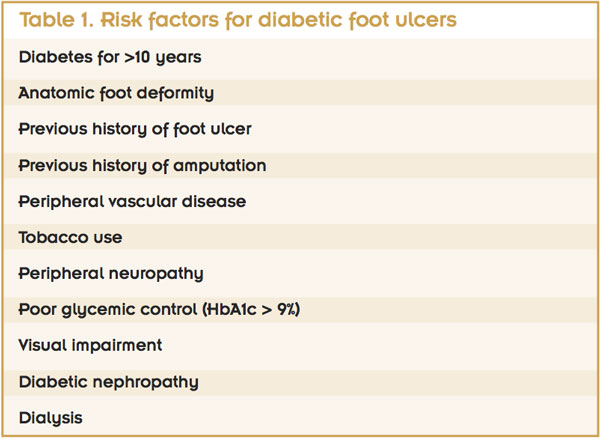
Risk factors for diabetic foot ulceration (DFU) can be categorized into three groups: pathophysiologic changes, anatomic deformities, and trauma.5 Pathophysiologic changes occur at the molecular level and lead to complications that include peripheral neuropathy, peripheral vascular disease, and a compromised immune system. These changes compromise wound-healing capabilities. Charcot neuroarthropathy and motor neuropathy are the major contributors to anatomic foot deformity, which in turn leads to increased peak plantar pressures and increased risk of skin breakdown. Finally, external influences in the form of acute or chronic trauma often are initiating factors in the development of diabetic foot ulcerations.
Additional risk factors include a long-term history of diabetes. Patients who have had diabetes for more than 20 years have a six-fold increase in the risk of foot ulceration compared to patients with a history of diabetes of nine years or less.6 Other significant known risk factors include a history of previous foot ulceration and a history of amputation.7

Figure 1. Pathway to foot ulceration.
It is important to understand that these risk factors do not typically occur independently, but rather in combination, further increasing the risk of ulceration. The pathway of diabetic foot ulceration often begins with an insensate foot. Then acute or chronic trauma occurs, often in an area of elevated peak plantar pressure, which leads to skin breakdown. Ulcer development can also be compounded by impaired wound healing due to reduced blood flow to the area and a lack of growth factors and cytokines, which prevents wound healing and closure.
Peripheral neuropathy
Pain is one of the body’s many normal responses to harmful stimuli. The sensation that ails so many of us is envied by millions of those diagnosed with diabetes across the world. This inability to detect the body’s warning signal of harm often initiates the pathway to foot ulceration in the insensate foot. It has been estimated that up to 85% of DFUs are associated with peripheral sensory neuropathy.8
Trauma
Trauma is also often an initiating factor of a DFU, and both acute and chronic trauma can contribute to their development. Typically trauma on the neuropathic foot occurs because of high pressures that develop during walking. Under normal circumstances, the foot is able to distribute these peak plantar forces, but this ability is impaired in those with diabetes, mainly because of foot changes due to motor neuropathy as well as limited joint mobility.9 Limited joint mobility is related to collagen glycosylation, which results in thickening of ligaments, tendons, and capsules. In one study, minor trauma, foot deformity, and peripheral neuropathy were found to be present in more than 63% of patients who developed DFUs.10
Anatomic deformity

Figure 2. A 76-year-old man with a history of diabetes and peripheral vascular disease with sub fifth metatarsal head ulceration that probes to bone and surrounding hyper- keratosis indicative of an area of peak plantar pressure.
Anatomic deformity is a leading contributing factor in the development of a diabetic ulceration.7,10 Diabetic autonomic and motor neuropathies are leading causes of foot deformities. These deformities lead to areas of increased peak plantar pressures, which are common sites of ulceration formation. It is important to understand the etiologies of these deformities in order to adequately manage and eliminate the deforming force.
Autonomic and motor neuropathy. The pathway to diabetic autonomic dysfunction is multifactorial, and clinical symptoms do not occur until long after the onset of diabetes. Subclinical symptoms however can occur within one to two years of onset.11 The etiology of diabetic autonomic and motor neuropathy is thought to be caused primarily by hyperglycemic activation of the polyol pathway, which in turn leads to direct neuronal damage through the accumulation of sorbitol and potential changes in the NAD:NADH (nicotinamide adenine dinucleotide: nicotinamide adenine dinucleotide phosphate) ratio. Reduction in neuronal blood flow via vasoconstriction occurs due to activation of protein kinase C.12
Figure 3a: A man with diabetes aged 47 years with advanced-stage Charcot deformity presented to an emergency department with an infected lateral malleolus ulcer after walking without his CROW (Charcot restraint orthotic walker) device. Note the bone exuding through the ulcer site and the presence of small maggots through- out. The patient went on to a below-the-knee amputation two days after this picture was taken.
Other causes of autonomic and motor neuropathy include advanced glycosylation end products that accumulate in the endoneural blood vessels that directly attach to and damage the nerve tissue.13 This leads to increased arterial dilation secondary to loss of vasoconstrictive mechanisms that are controlled by the central nervous system. Due to this dilation, the patient often has increased lower extremity neuropathic edema that is recalcitrant to diuretic therapy.14
Other common autonomic findings include increased warmth, erythema, or both due to dilation of the blood vessels. Furthermore, autonomic neuropathy decreases eccrine sweat gland function, leading to excessively dry skin.15 This xerotic skin change predisposes the patient to fissuring and subsequent ulceration. As discussed by Katoulis et al, patients with diabetes and subsequent peripheral neuropathy have some alterations in gait mechanics, which in turn may predispose the patient to foot injuries and foot ulcerations.16
The pathway to diabetic motor neuropathy, as described above, leads to wasting of the small intrinsic musculature in the foot, and to foot deformities.17 Common foot deformities that result from this type of neuropathy include hammertoes, which often lead to prominent metatarsal heads and corresponding increases in peak plantar pressures. Peak plantar pressures can occur anywhere on the foot where a deformity exists, and acute or chronic trauma to these areas puts patients at risk for skin breakdown.18
Charcot neuroarthropathy. The incidence of Charcot neuroarthropathy is estimated to be between .8% and 8% in people diagnosed with diabetes.19,20 Charcot neuroarthropathy is described by progressive joint dislocation, fracture, and resultant foot deformity, most commonly occurring in the midfoot region.21

Figure 3b: Computed tomography reconstruction of the same patient. Note the severe fragmentation of the ankle and tarsal bones.
From a pathophysiologic standpoint, two theories have been proposed that likely work in tandem. The neurotraumatic theory describes the process of repetitive joint microtrauma due to an insensate foot. The second theory is neurovascular, and is based upon autonomic neuropathy: The chronic dilation of blood vessels leads to increased osteoclastic activity and subsequent bone destruction and erosions.22-24 These two proposed etiologies may occur simultaneously, leading to subsequent bony deformity. These bony prominences lead to peak plantar pressures and thus skin breakdown. Sohn et al found that 59% of those with Charcot develop a foot ulceration due to the resultant deformity.23 That said, not all patients with Charcot develop a foot ulcer, and, therefore, it is important to focus on prevention of ulceration.
Peripheral vascular disease
While the association of DFUs with peripheral neuropathy is staggering, peripheral vascular disease (PVD) does not lag far behind. The reported incidence of PVD in DFUs is as high as 60%.21 It is important to note that peripheral neuropathy and PVD are not mutually exclusive, and most experts agree that typical DFUs are neuroischemic in nature (again, with many of the previously mentioned causal factors also contributing).25 It is critical to examine the unique nature of PVD in those diagnosed with diabetes, as it is characterized by impairment at both the macrovascular and microvascular level.
Macrovascular disease in those diagnosed with diabetes is similar to its manifestation in otherwise healthy patients. It is an occlusive disease that includes atherogenic changes to the coronary, carotid, and peripheral arterial walls. In nondiabetic patients, PVD is associated with risk factors such as smoking, hypertension and hyperlipidemia, and is more proximally based in the aorto-iliac-femoral-popliteal arteries. In contrast, PVD in patients with diabetes tends to involve below-knee tibial arteries in a more diffuse pattern with resultant poor collateral circulation.22 There is also a higher incidence of Monckeberg’s sclerosis, or calcification of the intimal plaque and media, with macrovascular diabetic disease.26
Microvascular disease relates primarily to capillaries and arterioles. Microvascular disease in a patient with diabetes is not an occlusive phenomenon, as it was once thought to be. Hyperglycemia and insulin resistance produce both structural and functional changes within the arteriolar and capillary levels.27 Structurally, there is a marked thickening of the basement membrane and reduction in capillary size. Interestingly, in patients with type 1 diabetes, researchers have found a direct correlation between the degree of basement membrane thickening and glycemic control.28

These structural changes affect numerous cellular functions and ultimately result in increased vascular permeability and impaired autoregulation of blood flow and vascular tone. Consequently, the normal hyperemic response to injury is impaired and reduced. This translates into suboptimal perfusion at the capillary level in DFUs, which so desperately need blood flow to heal. Decreased capillary elasticity, impaired cellular migration, and impaired nutrient exchange are a few of the more detrimental functional consequences of microvascular dysfunction in individuals with diabetes.
Overt microvascular complications include proliferative retinopathy, nephropathy, and neuropathy. One study investigated trends in the prevalence of these early microvascular complications over a 17-year period (1990-2006) in adolescents with type 1 diabetes. Investigators found that the prevalence of early retinopathy declined from 1990 to 2002, then remained unchanged until 2006. Albumin excretion rate elevation (7.5 µg/min or greater) and microalbuminuria (20 µg/min or greater) did not change over time, but peripheral nerve abnormalities increased during the studied period despite advances in education and technology.29
 Prostaglandin transporter (PGT) is an important mediator of prostaglandin catabolism and signal termination. In those with diabetes, the prostaglandin PGE(2) is diminished in skin; this normally induces angiogenesis and vasodilation. A recent study in mice with diabetes targeted this concept and found that pharmacologic inhibition of PGT corrected this imbalance and resulted in shorter times to wound closure. Investigators concluded that hyperglycemia enhances PGT expression, which in turn diminishes angiogenic-signaling pathways that may be a critical mechanism for wound healing in DFUs.30
Prostaglandin transporter (PGT) is an important mediator of prostaglandin catabolism and signal termination. In those with diabetes, the prostaglandin PGE(2) is diminished in skin; this normally induces angiogenesis and vasodilation. A recent study in mice with diabetes targeted this concept and found that pharmacologic inhibition of PGT corrected this imbalance and resulted in shorter times to wound closure. Investigators concluded that hyperglycemia enhances PGT expression, which in turn diminishes angiogenic-signaling pathways that may be a critical mechanism for wound healing in DFUs.30
Impaired wound healing
Wound healing is a complex process that involves connective tissue formation, cellular activity, and growth factor activation. Those diagnosed with diabetes have alterations of all three of these physiologic processes.31 Collagen is the most abundant protein in connective tissue, and the balance of its synthesis and degradation is essential for normal wound repair. In the diabetic state, this tenuous balance can shift, disrupting the wound healing process.
In normal wound repair, the inflammatory stage typically lasts two to three days. This stage involves a well-orchestrated interaction among various cells—such as epithelial cells, fibroblasts, dendritic cells, and endothelial cells—with biochemical activity. Neutrophils, T-cells, natural killer cells, macrophages, and platelets also are attracted and recruited to the wound to mediate inflammation, coagulation, and angiogenesis processes. Stojadinovic et al referred to the DFU as being “stuck” in this stage, as altered secretion and aberrant production of growth factors are noted.31
Growth factors exhibit their wound healing effects by either inhibition or stimulation of the local environment. Growth factors such as platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), and vascular endothelial growth factor (VEGF) are all critical in chemotaxis, migration, stimulation, and proliferation of cells and matrix substances for wound healing.
A recent study by Dinh et al looked at how vascular function and inflammation relate to the development and healing patterns of DFUs. In the patients with DFUs that did not heal (47%), elevated serum levels of tumor necrosis factor-α, monocyte chemoattractant protein-1, matrix metallopeptidase 9 (MMP-9), and fibroblast growth factor were observed when compared to the 53% of patients with DFUs that did heal.32
Dinh et al also analyzed skin biopsy specimens and found that patients with diabetes had increased immune cell infiltration, expression of MMP-9, and protein tyrosine phosphatase-1B (PTP1B). PTP1B has been shown to negatively regulate insulin, leptin, and various growth factor signaling pathways. The authors concluded that the irregular growth factor levels and increased expression of MMP-9 and PTP1B are integral to poor healing in DFUs.32 Therefore, it would make sense to target treatments at inhibiting PTP1B, MMP-9, or both. Currently, there are local PTP1B inhibitors that are under consideration as a possible treatment for type 2 diabetes.33
Conclusion
With diabetes incidence at an all-time high and continuing to rise, understanding the risk factors responsible for development of diabetic foot complications is critical to proper treatment.
Clearly, the etiology of diabetic foot ulcers is complex and multifactorial, requiring a multidisciplinary approach to address the numerous deficiencies, which range from overt foot deformities to a diminished healing capacity at the microscopic level. In this review, we have briefly examined the risk factors currently known to be instrumental in development of diabetic foot complications. However, our understanding continues to grow as research on the diabetic foot expands.
Allyson Berglund, DPM, and Matthew Juriga, DPM, are podiatric surgical residents (PGY-2) at Beth Israel Deaconess Medical Center and clinical fellows in surgery at Harvard Medical School in Boston. Aristidis Veves, MD, DSc, is associate professor of surgery at Harvard Medical School and research director of the Microcirculation Lab at Joslin-Beth Israel Deaconess Foot Center in Boston. Thanh Dinh, DPM, is a clinical instructor in surgery at Harvard Medical School and Beth Israel Deaconess Medical Center.
- Wild S, Roglic G, Green A, et al. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 2004;27(5):1047-1053.
- Brem H, Tomic-Canic M. Cellular and molecular basis of wound healing in diabetes. J Clin Invest 2007;117(5):1219–1222.
- Reiber GE, Boyko EJ, Smith DG. Lower extremity foot ulcers and amputations in diabetes. In: Harris MI, Cowie CC, Stern MP, et al, eds. Diabetes in America. 2nd ed. Washington DC: US Govt Printing Office; 1995:409-428.
- Boyko EJ, Ahroni JH, Smith DG, Davignon D. Increased mortality associated with diabetic foot ulcer. Diabet Med 1996;13(11):967-972.
- Dinh TL, Veves A. A review of the mechanisms implicated in the pathogenesis of the diabetic foot. Int J Lower Extrem Wounds 2005;4(3):154-159.
- Rith-Najarian SJ, Stolusky T, Gohdes DM. Identifying diabetic patients at high risk for lower-extremity amputation in a primary health care setting. Diabetes Care 1992;22(10):1386–1389.
- Boyko EJ, Ahroni JH, Stensel V, et al. A prospective study of risk factors for diabetic foot ulcer: the Seattle Diabetic Foot Study. Diabetes Care 1999;22(7):1036-1042.
- Ndip A, Ebah L, Mbako A. Neuropathic diabetic foot ulcers–evidence-to-practice. Int J Gen Med 2012;5:129-134.
- Dinh TL, Veves A. The diabetic foot. In: Defronzo RA, Ferrannini E, Keen H, Zimmet P, eds. International Textbook of Diabetes Mellitus. 3rd ed. Chichester, West Sussex, UK: John Wiley & Sons, Ltd; 2004:1315-1332.
- Reiber GE, Vileikyte L, Boyko EJ, et al. Causal pathways for incident lower-extremity ulcers in patients with diabetes from two settings. Diabetes Care 1999;22(1):157–162
- Pfeifer MA, Weinberg CR, Cook DL, et al. Autonomic neural dysfunction in recently diagnosed diabetic subjects. Diabetes Care 1984;7(5):447-453.
- Veves A, King GL. Can VEGF reverse diabetic neuropathy in human subjects? J Clin Invest 2001;107(10):1215-1218.
- Brownlee M. Glycation products and the pathogenesis of diabetic complications. Diabetes Care 1992;15(12):1835-1843.
- Ward JD, boulton AJ, Simms JM, et al. Venous distension in the diabetic neuropathic foot. J R Soc Med 1983;76(12):1011-1014.
- Petrofsky J, Berk L, Al-Nakhli H. The influence of autonomic dysfunction associated with aging and type 2 diabetes on daily life activities. Exp Diabetes Res 2012;2012:657103.
- Katoulis EC, Ebdon-Parry M, Lanshammar H, et al. Gait abnormalities in diabetic neuropathy. Diabetes Care 1997;20(12):1904-1907.
- Andersen H. Motor dysfunction in diabetes. Diabetes Metab Rev 2012;28(Suppl 1):89-92.
- Boulton AJ. The importance of abnormal foot pressures in early diabetic neuropathy. Diabetic Med 1987;4(3):225-228.
- Rajbhandari SM, Jenkins RC, Davies C, Tesfaye S. Charcot neuroarthropathy in diabetes mellitus. Diabetologia 2002;45(8):1085-1096.
- Fabrin J, Larsen K, Holstein PE. Long-term follow-up in diabetic Charcot feet with spontaneous onset. Diabetes Care 2000;23(6):796-800.
- Trepman E, Nihal A, Pinzur MS. Current topics review: Charcot neuroarthropathy of the foot and ankle. Foot Ankle Int 2005;26(1):46-63.
- Larson SA, Burns PR. The pathogenesis of Charcot neuroarthropathy: current concepts. Diabet Foot Ankle 2012;3:12236.
- Sohn MW, Lee TA, Stuck RM, et al. Mortality risk of Charcot arthropathy compared with that of diabetic foot ulcer and diabetes alone. Diabetes Care 2009;32(5):816-821.
- Wukich DK, Sung W. Charcot arthropathy of the foot and ankle: modern concepts and management review. J Diabetes Complications 2009;23(6):409-426.
- Ndip A, Jude EB. Emerging evidence for neuroischemic diabetic foot ulcers: model of care and how to adapt practice. Int J Low Extrem Wounds 2009;8(2):82-94.
- Gibbons GW, Shaw PM. Diabetic vascular disease: characteristics of vascular disease unique to the diabetic patient. Semin Vasc Surg 2012;25(2):89-92
- Tecilazich F, Dinh T, Kafanas A, Veves A. Microvascular changes in the diabetic foot. In: Veves A, Giurini JM, LoGerfo FW, eds. The Diabetic Foot: Medical and Surgical Management. 3rd ed. New York, NY: Humana Press; 2012:185-201.
- Raskin P, Pietri AO, Unger R, Shannon WA Jr. The effect of diabetic control on the width of skeletal-muscle capillary basement membrane in patients with type I diabetes mellitus. N Engl J Med 1983;309(25):1546-1550.
- Cho YH, Craig ME, Hing S, et al. Microvascular complications assessment in adolescents with 2- to 5-yr duration of type 1 diabetes from 1990 to 2006. Pediatr Diabetes 2011;12(8):682-689.
- Syeda MM, Jing X, Mirza RH, et al. Prostaglandin transporter modulates wound healing in diabetes by regulating prostaglandin-induced angiogenesis. Am J Pathol 2012;181(1):334-346.
- Stojadinovic O, Pastar I, Gordon KA, et al. Physiology and pathophysiology of wound healing in diabetes. In: Veves A, Giurini JM, LoGerfo FW, eds. The Diabetic Foot: Medical and Surgical Management. 3rd ed. New York, NY: Humana Press; 2012:127-150.
- Dinh T, Tecilazich F, Kafanas A, et al. Mechanisms involved in the development and healing of diabetic foot ulceration. Diabetes 2012;61(11):2937-2947.
- Erbe DV, Klaman LD, Wilson DP, et al. Prodrug delivery of novel PTP1B inhibitors to enhance insulin signaling. Diabetes Obes Metab 2009;11(6):579-588.